

Description
Hemophilia is a bleeding disorder that slows the blood clotting process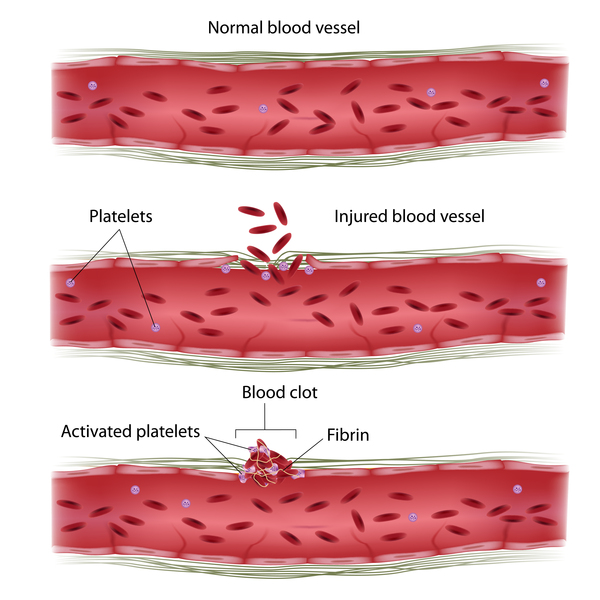 . People with this condition experience prolonged bleeding or oozing following an injury, surgery, or having a tooth pulled. In severe cases of hemophilia, continuous bleeding occurs after minor trauma or even when there is no obvious injury (sometimes called spontaneous bleeding). Serious complications can result from bleeding into the joints, muscles, brain, or other internal organs. Milder forms of hemophilia do not necessarily involve spontaneous bleeding, and the condition may not become apparent until abnormal bleeding occurs following surgery or a serious injury.
. People with this condition experience prolonged bleeding or oozing following an injury, surgery, or having a tooth pulled. In severe cases of hemophilia, continuous bleeding occurs after minor trauma or even when there is no obvious injury (sometimes called spontaneous bleeding). Serious complications can result from bleeding into the joints, muscles, brain, or other internal organs. Milder forms of hemophilia do not necessarily involve spontaneous bleeding, and the condition may not become apparent until abnormal bleeding occurs following surgery or a serious injury.
The major types of this condition are hemophilia A (also known as classic hemophilia or factor VIII deficiency) and hemophilia B (also known as Christmas disease or factor IX deficiency). Although the two types have very similar signs and symptoms, they are caused by variants (also known as mutations) in different genes. People with an unusual form of hemophilia B, known as hemophilia B Leyden, experience episodes of excessive bleeding in childhood but have few bleeding problems after puberty.
Frequency
The two major forms of hemophilia occur much more commonly in males than in females. Hemophilia A is the most common type of the condition; 1 in 4,000 to 1 in 5,000 males worldwide are born with this disorder. Hemophilia B occurs in approximately 1 in 20,000 newborn males worldwide.
Causes
Variants in the F8 gene cause hemophilia A, while variants in the F9 gene cause hemophilia B. The F8 gene provides instructions for making a protein called coagulation factor VIII. A related protein, coagulation factor IX, is produced from the F9 gene. Coagulation factors are proteins that work together in the blood clotting process. After an injury, blood clots protect the body by sealing off damaged blood vessels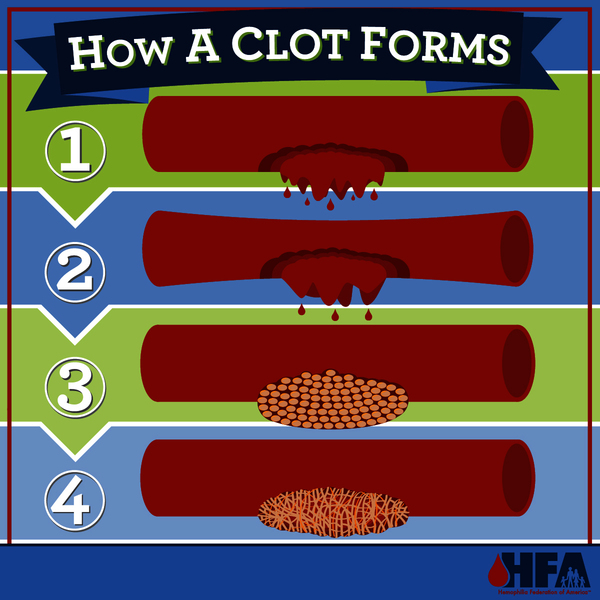 and preventing excessive blood loss.
and preventing excessive blood loss.
Variants in the F8 or F9 gene lead to the production of an abnormal version of coagulation factor VIII or coagulation factor IX, or reduce the amount of one of these proteins. The altered or missing protein cannot participate effectively in the blood clotting process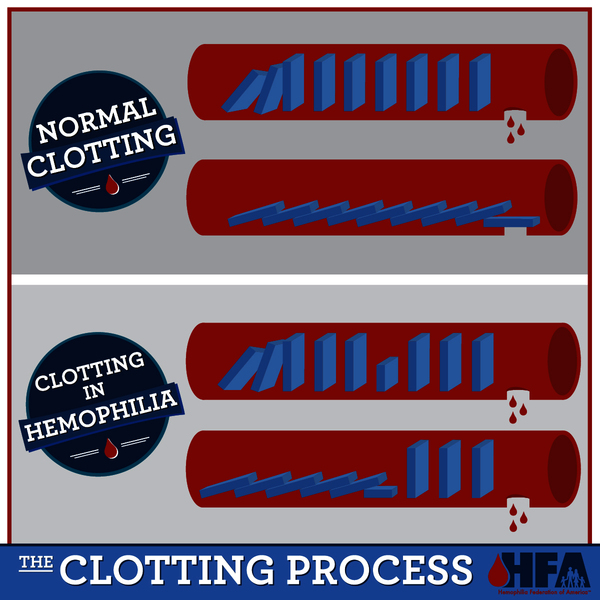 . As a result, blood clots cannot form properly in response to injury. These problems with blood clotting lead to continuous bleeding that can be difficult to control. The variants that cause severe hemophilia almost completely eliminate the activity of coagulation factor VIII or coagulation factor IX. The variants involved in mild and moderate hemophilia reduce but do not eliminate the activity of one of these proteins.
. As a result, blood clots cannot form properly in response to injury. These problems with blood clotting lead to continuous bleeding that can be difficult to control. The variants that cause severe hemophilia almost completely eliminate the activity of coagulation factor VIII or coagulation factor IX. The variants involved in mild and moderate hemophilia reduce but do not eliminate the activity of one of these proteins.
Another form of the disorder, known as acquired hemophilia, is not caused by inherited gene variants. This rare condition is characterized by abnormal bleeding into the skin, muscles, or other soft tissues, usually beginning in adulthood. Acquired hemophilia results when the body makes specialized proteins called autoantibodies that attack and disable coagulation factor VIII. The production of autoantibodies is sometimes associated with pregnancy, immune system disorders, cancer, or allergic reactions to certain drugs. In about half of cases, the cause of acquired hemophilia is unknown.
Inheritance
Hemophilia A and hemophilia B are inherited in an X-linked recessive pattern . The genes associated with these conditions are located on the X chromosome, which is one of the two sex chromosomes
. The genes associated with these conditions are located on the X chromosome, which is one of the two sex chromosomes . In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
. In males (who have only one X chromosome), one altered copy of the gene in each cell is sufficient to cause the condition. A characteristic of X-linked inheritance is that fathers cannot pass X-linked traits to their sons.
In females (who have two X chromosomes), a variant would usually have to occur in both copies of the gene to cause the disorder. However, in some instances, one altered copy of the F8 or F9 gene is sufficient, because the X chromosome with the normal copy of the gene is turned off through a process called X-inactivation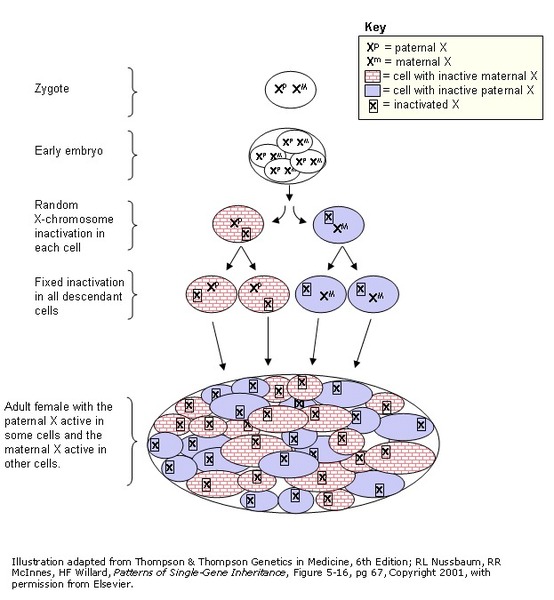 . X-inactivation occurs early in embryonic development in females. Through this process, one of the two X chromosomes is permanently turned off (inactivated) in somatic cells (cells other than egg and sperm cells). X-inactivation ensures that females, like males, have only one active copy of the X chromosome in each body cell.
. X-inactivation occurs early in embryonic development in females. Through this process, one of the two X chromosomes is permanently turned off (inactivated) in somatic cells (cells other than egg and sperm cells). X-inactivation ensures that females, like males, have only one active copy of the X chromosome in each body cell.
Usually X-inactivation occurs randomly, such that each X chromosome is active in about half of the body cells. Sometimes X-inactivation is not random, and one X chromosome is active in more than half of cells. When X-inactivation does not occur randomly, it is called skewed X-inactivation.
In many females with a variant in one copy of the F8 or F9 gene, X-inactivation is random and the chromosome with the normal copy of the gene is turned off in about half of cells. These individuals have about half the usual amount of coagulation factor VIII or coagulation factor IX, which is generally enough for normal clotting. However, in some females with an F8 or F9 gene variant, X-inactivation is skewed, and the chromosome with the normal copy of the gene is turned off in more than half of cells. These individuals can have less coagulation factor VIII or coagulation factor IX than usual and are at risk of abnormal bleeding.
Other Names for This Condition
- Haemophilia
- Hemophilia, familial
- Hemophilia, hereditary
Additional Information & Resources
Genetic and Rare Diseases Information Center
Patient Support and Advocacy Resources
Clinical Trials
Catalog of Genes and Diseases from OMIM
Scientific Articles on PubMed
References
- Bolton-Maggs PH, Pasi KJ. Haemophilias A and B. Lancet. 2003 May 24;361(9371):1801-9. doi: 10.1016/S0140-6736(03)13405-8. Citation on PubMed
- Franchini M. Acquired hemophilia A. Hematology. 2006 Apr;11(2):119-25. doi: 10.1080/10245330600574185. Citation on PubMed
- Giangrande P. Haemophilia B: Christmas disease. Expert Opin Pharmacother. 2005 Aug;6(9):1517-24. doi: 10.1517/14656566.6.9.1517. Citation on PubMed
- Graw J, Brackmann HH, Oldenburg J, Schneppenheim R, Spannagl M, Schwaab R. Haemophilia A: from mutation analysis to new therapies. Nat Rev Genet. 2005 Jun;6(6):488-501. doi: 10.1038/nrg1617. Citation on PubMed
- Konkle BA, Nakaya Fletcher S. Hemophilia A. 2000 Sep 21 [updated 2023 Jul 27]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1404/ Citation on PubMed
- Konkle BA, Nakaya Fletcher S. Hemophilia B. 2000 Oct 2 [updated 2023 Feb 9]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from http://www.ncbi.nlm.nih.gov/books/NBK1495/ Citation on PubMed
- McLintock C. Women with bleeding disorders: Clinical and psychological issues. Haemophilia. 2018 May;24 Suppl 6:22-28. doi: 10.1111/hae.13501. No abstract available. Citation on PubMed
- Oldenburg J, El-Maarri O. New insight into the molecular basis of hemophilia A. Int J Hematol. 2006 Feb;83(2):96-102. doi: 10.1532/IJH97.06012. Citation on PubMed
- Plug I, Mauser-Bunschoten EP, Brocker-Vriends AH, van Amstel HK, van der Bom JG, van Diemen-Homan JE, Willemse J, Rosendaal FR. Bleeding in carriers of hemophilia. Blood. 2006 Jul 1;108(1):52-6. doi: 10.1182/blood-2005-09-3879. Epub 2006 Mar 21. Citation on PubMed
- van Galen KPM, d'Oiron R, James P, Abdul-Kadir R, Kouides PA, Kulkarni R, Mahlangu JN, Othman M, Peyvandi F, Rotellini D, Winikoff R, Sidonio RF. A new hemophilia carrier nomenclature to define hemophilia in women and girls: Communication from the SSC of the ISTH. J Thromb Haemost. 2021 Aug;19(8):1883-1887. doi: 10.1111/jth.15397. Citation on PubMed
The information on this site should not be used as a substitute for professional medical care or advice. Contact a health care provider if you have questions about your health.